

Elternerklärung
Erstellt in Zusammenarbeit mit :
Prof. Dr. Christian Körner, Prof. Dr. Georg. F. Hoffmann, Dr. Christian Thiel
Universitätsklinik für Kinder- und Jugendmedizin Heidelberg
Wie kann man sich die Ursachen für CDG innerhalb der Körperzellen vorstellen ?
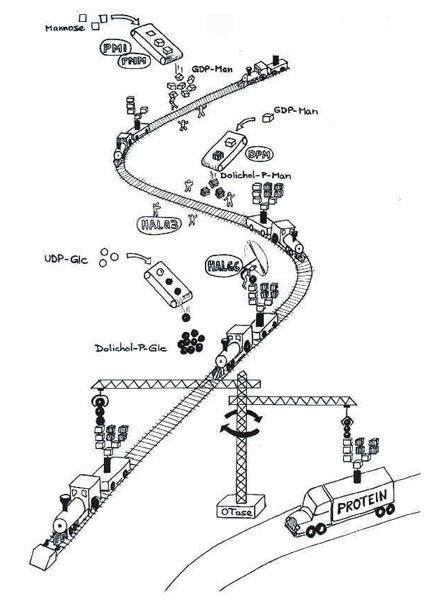
Jedes Organ in unserem Körper ist aus vielen Millionen von Zellen aufgebaut, die unterschiedliche Aufgaben haben. So sollen beispielsweise die Magenzellen Verdauungsstoffe zur Verarbeitung der Nahrung herstellen oder die Zellen des Auges all das produzieren, was man zum Sehen benötigt. Bildlich betrachtet gibt es in jeder Zelle eine komplexe Fabrik (siehe Abbildung 3) mit mehreren Arbeitsbereichen, wie das Cytosol, das endoplasmatische Retikulum und den Golgi-Apparat (siehe Abbildung 2). Unter den Produkten, die in der Fabrik hergestellt werden, sind sehr viele Proteine (Eiweiße), die unser Körper benötigt. Damit viele dieser Proteine allerdings so funktionieren können wie sie sollen, müssen sie in der Fabrik noch mit Zuckerketten verbunden werden. Diese Ketten werden zunächst aus 14 einzelnen Zuckermolekülen zusammengesetzt, wobei der größte Teil aus einem Verwandten des Traubenzuckers besteht, den man Mannose nennt.
{kind=link}
{kind=link}
Jeder einzelne Zucker wird durch einen eigenen spezialisierten Arbeiter (Enzym) in der Fabrik eingebaut. Später werden die so entstandenen Zuckerketten auf Proteine übertragen und dann durch andere Enzyme weiter bearbeitet. So entstehen nach mehr als vierzig Einzelschritten Zuckereiweißkörper, die so genannten Glykoproteine. Tritt in der Fabrik ein Produktionsfehler auf (bedingt durch eine Störung in einem spezifischen Produktionsschritt, z.B. durch einen Enzymdefekt), können die Zuckerketten an den einzelnen Proteinen nicht korrekt gebildet werden. Dies führt zu einem Funktionsverlust der Proteine und damit zu CDG.
Abbildung 3 stellt die Herstellung der Zuckerketten, die später an Proteine geknüpft werden, innerhalb von Zellen dar. Die Zuckerketten (auf dem Zugwagen) werden durch schrittweise Zugabe von Zucker-Bausteinen aufgebaut. Diese Bausteine heißen z.B. GDP-Mannose (GDP-Man), Dolichol-Phosphat-Mannose (Dolichol-P-Man), und Dolichol-Phosphat-Glucose (Dolichol-P-Glc). Die Arbeiter (Enzyme) entlang der Eisenbahn bereiten die Bausteine vor und übertragen sie auf die Zuckerketten. Wenn die Ketten fertig gestellt sind, werden sie vom Oligosaccharyl-Transferase-Komplex (OTase Kran) zu den Proteinen (Lastwagen) transportiert und übertragen. Noch während sich das Protein in seine richtige Form faltet, werden die Glucosereste sowie ein Mannoserest von den Enzymen Glucosidase und Mannosidase wieder abgespalten. Anschließend wird das Protein in den Golgi-Apparat transportiert, wo die Zuckerketten zunächst weiter verkürzt und anschließend in einer genau festgelegten Reihenfolge um weitere Zuckerreste (N-Acetylglucosamin, Galaktose, Fucose, Sialinsäure) verlängert werden (siehe Abbildung 2).
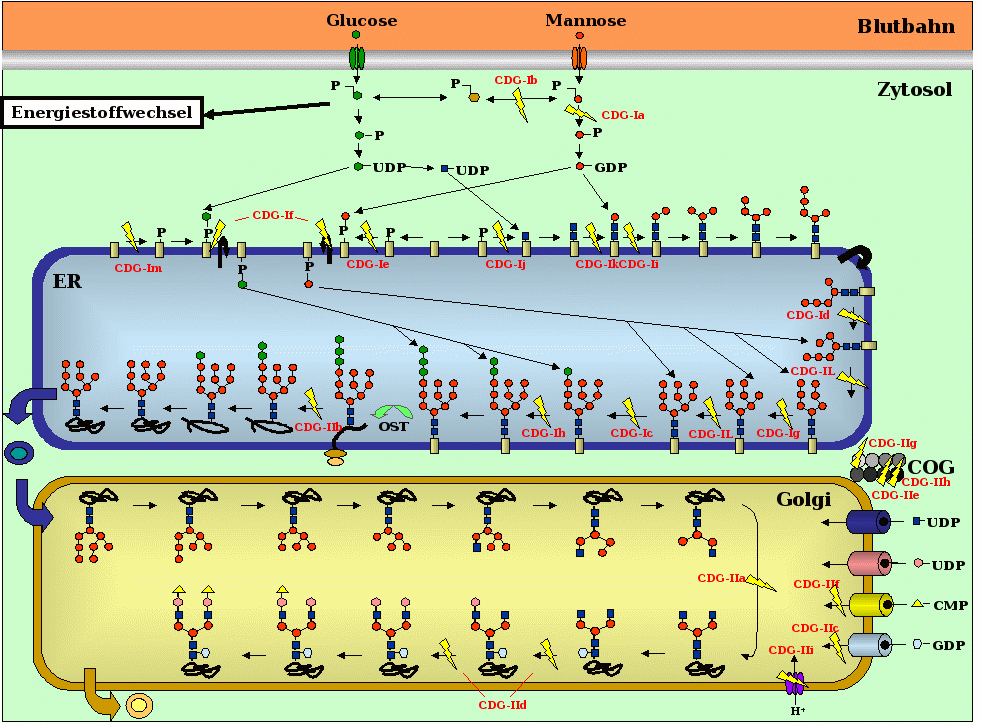
Eine Glykosylierungsstörung, die zu CDG führt, kann durch defekte Enzyme, Transporter, Transportsysteme oder eine verminderte Bereitstellung der einzelnen Zuckerbausteine verursacht werden (siehe Abbildung 2). Dabei umfassen CDG-1 alle Defekte, die im Cytosol sowie am und im endoplasmatischen Retikulum (ER) bis zum Transfer des Oligosaccharids durch die Oligosaccharyltransferase auf das Protein auftreten. Alle nachfolgenden Defekte werden unter CDG-2 zusammengefasst. Weiterhin erhalten alle neuen Defekte, deren molekulare Ursache aufgeklärt worden ist, einen Buchstaben nach fortlaufendem Alphabet. Defekte, deren Identifikation der molekularen Ursache noch aussteht, werden als CDG-1x oder CDG-2x bezeichnet.
Im Folgenden werden die molekularen Ursachen für die bisher bekannten CDG-Typen kurz beschrieben, wobei man sich an der Abbildung 2 orientieren kann.
Bei CDG-1a und CDG-1b wird der Baustein GDP-Man nur in unzureichender Menge im Cytosol gebildet. Bei Patienten mit CDG-1c und CDG-Ih funktioniert die Übertragung von Glucose im endoplasmatischen Retikulum aufgrund von defekten Glucosyltransferasen nicht. CDG-1e Patienten synthetisieren zu wenig Dolichol-P-Man. Bei CDG-1f werden zu wenig Dolichol-P-Man und Dolichol-P-Glc für die Übertragungen von Mannose bzw. Glucose im Inneren des endoplasmatischen Retikulums für die Verlängerungen der Zuckerketten bereitgestellt. Die Ursachen für die CDG-Typen 1d, 1g, 1i, 1k und 1L beruhen auf der unzureichenden Übertragung von Mannoseresten, die durch die verminderte Aktivität von Mannosyltransferasen auf der äußeren bzw. inneren Seite des endoplasmatischen Retikulums hervorgerufen wird. Im Fall von CDG-1j-Patienten können N-Acetylglucosaminreste nicht in ausreichender Menge auf Dolicholphosphat übertragen werden. Bei CDG-2a und CDG-2d handelt es sich um Erkrankungen, bei denen die Übertragung der Zucker N-Acetylglucosamin bzw. Galactose aufgrund defekter Glycosyltransferasen im Golgi-Apparat beeinträchtigt ist. Die Ursache für CDG-2b konnte in einem Defekt des Enzyms Glucosidase I lokalisiert werden, wodurch die Patienten nicht in der Lage sind, den äußeren Glucoserest von vollständig gebildeten Zuckerkette wieder abzuspalten, was eine der Voraussetzungen für den Transport von Glykoproteinen in den Golgi-Apparat ist. Bei CDG-2c und CDG-2f handelt es sich um Defekte von Zuckertransportern, die für den Import von Fucose bzw. Sialinsäure in den Golgi-Apparat verantwortlich sind. CDG-2e und CDG-2g-Patienten sind von einem defekten Proteinkomplex betroffen, der vermutlich an der Anordnung von Glykosyltransferasen im Golgi-Apparat beteiligt ist.
Adresse | Ansprechpartner
| E-Mail:Bundesverein [at] cdg-syndrom.de | |
Spendenkonto
Kreissparkasse Esslingen-Nürtingen
IBAN DE89 6115 0020 0101 9723 05
BIC: ESSLDE66XXX
Amtsgericht Duisburg -
Vereinsregisterblatt 6282
Hinweis: Bitte geben Sie bei Spenden Ihre vollständige Anschrift in der Überweisung an, damit wir Ihnen eine Spendenbescheinigung zusenden können. Alternativ können Sie das Spenden-Formular >HIER< benutzen. Die Spendenbescheinigungen werden zu Beginn des folgenden Jahres verschickt.