

Was sind Congenital Disorders of Glycosylation (CDG) ?
Erstellt in Zusammenarbeit mit :
Prof. Dr. Christian Körner, Prof. Dr. Georg. F. Hoffmann, Dr. Christian Thiel,
Universitätsklinik für Kinder- und Jugendmedizin Heidelberg
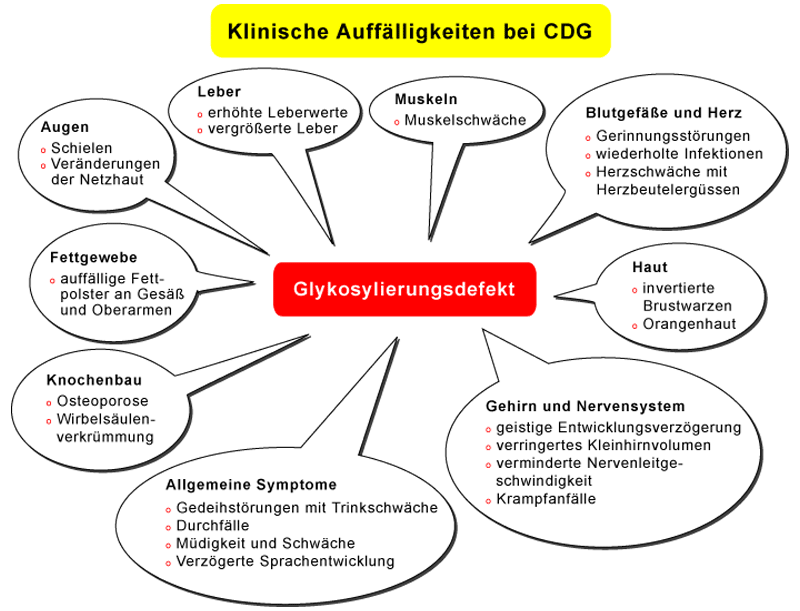
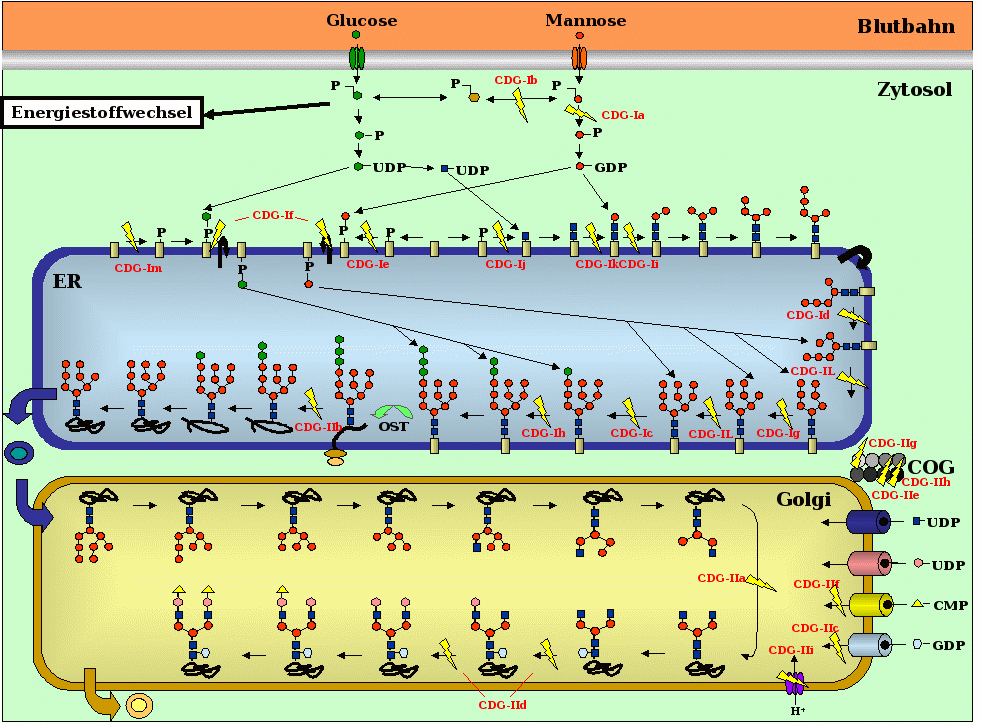
CDG, aus dem Englischen übersetzt `Erblich bedingte Erkrankungen der Glykosylierung´, zählen zur großen Gruppe der genetisch-bedingten Stoffwechseldefekte und damit überlappend auch zur Gruppe der Orphan-Erkrankungen bzw. seltenen Erkrankungen. Obwohl CDG häufig mit schweren, verschiedene Organe betreffenden Symptomen einhergehen (siehe Abbildung 1), wurden die ersten beiden Patienten erst vor etwa 25 Jahren von dem belgischen Professor für Kinderheilkunde Jaak Jaeken beschrieben. Seither gelang es die molekularen Ursachen von neunzehn verschiedenen CDG-Typen aufzuklären, die in zwei Hauptgruppen unterteilt werden (CDG-I und CDG-II). Die Eingruppierung erfolgt nach der Lokalisation des jeweiligen Defektes innerhalb der Zelle und nicht nach klinischen Gesichtspunkten (siehe Abbildung 2). Die verschiedenen CDG-Typen können mit sehr unterschiedlichen Symptomen und Krankheitsverläufen einhergehen und werden daher oftmals spät oder gar nicht erkannt.
{kind=link}
{kind=link}
Sekundäre Glykosylierungsstörungen, beispielsweise bei den Stoffwechselerkrankungen Galaktosämie und Fructoseintoleranz oder bei Alkoholabusus, müssen bei der Untersuchung auf CDG ausgeschlossen werden.
Adresse | Ansprechpartner
| E-Mail:Bundesverein [at] cdg-syndrom.de | |
Spendenkonto
Kreissparkasse Esslingen-Nürtingen
IBAN DE89 6115 0020 0101 9723 05
BIC: ESSLDE66XXX
Amtsgericht Duisburg -
Vereinsregisterblatt 6282
Hinweis: Bitte geben Sie bei Spenden Ihre vollständige Anschrift in der Überweisung an, damit wir Ihnen eine Spendenbescheinigung zusenden können. Alternativ können Sie das Spenden-Formular >HIER< benutzen. Die Spendenbescheinigungen werden zu Beginn des folgenden Jahres verschickt.